

_-The-Breakthrough-Antipsychotic-That-Could-Change-Everything.webp?t=1729528747 "KarXT (Cobenfy)_ The Breakthrough Antipsychotic That Could Change Everything.jpg")
Learning Objectives
After the webinar, clinicians should:
1. Describe what black box warning means
2. Explain when black box warnings are indicated
3. Develop competency in assessment of the risks associated with boxed warnings for antidepressants
4. Understand how to assess for and document these risks
I'm here to talk to you about black box warning in children and adolescents, part of a Carlat webinar. I am a psychiatric nurse practitioner, and I work at John Peter Smith Hospital, part of Acclaim Multi-Specialty Group in Fort Worth, Texas. I have no conflicts or disclosures.
After the webinar, clinicians should describe what black box warning means, explain when black box warnings are indicated, develop competency in assessment of the risks associated with black box warnings for antidepressants, and understand how to assess for and document these risks.
[Transcript edited for clarity]
Technically, a black box warning is a boxed warning that happens to be outlined in black, so there is no need to say in your documentation or to parents that you've discussed the black box warning—you can just write “boxed warning.”
The basics
Let's get into the basics for the beginnings of the boxed warning. They were first introduced in 1979. They are the highest, most stringent safety-related warning that medications can be assigned by the FDA. There are over 500 different medications currently with boxed warnings. Typically, they are applied to a drug class rather than one specific drug. Warnings can be added, edited, or retracted over time.
Black box warnings are decided when a drug is being developed, with a plan to start when the drug is approved. However, more often than not, warnings occur after the drug has been approved based on what is called “post-marketing safety information or surveillance,” such as adverse reactions reported by consumers or healthcare professionals like you or me.
What is the approval process for a drug through the FDA? It's a very intensive process for a drug to even get to the stages of being approved. You can see on the right-hand side all of the stages. You'll probably be familiar with Phase 1 trials, where there are a small number of volunteers; Phase 2 with several hundred patients; and then Phase 3 where we start to get to several thousand patients who are taking the drug.
And during this process, there's a multidisciplinary team of chemists, pharmacologists, statisticians, and medical officers who carefully review the scientific information and with the sponsor negotiate labeling for a specific drug.
Who decides black box warnings are needed?
One place is through MedWatch, which is where providers and consumers report adverse side effects. There's also the adverse event reporting system, which was established in 1969 where the MedWatch cases are gathered and reviewed by pharmacists. If there's an adverse effect that is significant, it can create what's called a signal. And this leads specifically trained pharmacists to then find additional cases in the database and medical literature or even information from foreign regulatory agencies on that adverse effect
Sometimes they'll even have outside investigators who have access to large population-based databases to look to see if there were adverse effects there. And lastly, there can be advisory committees of experts who will present recommendations to the FDA. And typically, in those cases, when the committees come together, the FDA will follow their recommendation.
How do providers get notified of changes?
There are three main ways. Firstly, medication guides are available at pharmacies. Secondly, you can go to the FDA website. They will have advisories that are posted on potential safety issues that are identified even before the boxed warning is added. Lastly, prescribers can be notified through dear prescriber letters that are sent out to them.
Over-the-counter versus prescription drugs
The FDA regulates the advertising for prescription drugs and can mandate warnings like the ones you see at the end of commercials, where they speak really fast and go through a lot of different things. Over-the-counter advertisements are mandated by the FTC, the Federal Trade Commission. An interesting thing to take into account is that the FDA requires a warning on all prescriptions containing acetaminophen that overdose can result in liver transplant or death. So a lot of this could be medications that have even a really small dose of acetaminophen; even 325 mg, they'll have this warning.
The interesting part is that over-the-counter meds do have precautions, but they don't have a boxed warning. Now in both cases, whether a prescription or over the counter, they can lead to fatality or liver transplant, so it's a reminder that even over the counter medications that we may prescribe or not think too much about in terms of the risks, we really should be thinking about and teaching parents and kids about the potential side effects.
Boxed warnings are common. Changes are, too
In a JAMA study of 222 drugs approved between 2001 and 2010, researchers found manufacturers pulled three drugs off the market and the FDA required 61 black box warnings. Overall, black box warnings have greatly increased in the 2000s compared to earlier times. Even a study in 2006 found that more than 40% of patients over a 30-month span were prescribed at least one medication with a boxed warning. The point of all of this is to say that we are seeing a lot of patients who are prescribed medications with boxed warnings, and we're also seeing changes to whether a medication has a boxed warning or doesn't have one over time.
When are boxed warnings indicated?
Well, there are a number of reasons boxed warnings come about, but let's go over three global reasons and some examples.
1. First, when an adverse reaction is so serious in comparison to the drug’s benefits, a risk/benefit needs to be assessed. This would be for disabling or even fatal reactions. An example of this is the risk of anaphylaxis with iron dextran injection justifies its use only when anemia is severe and refractory to oral therapy. Essentially, there are some potent side effects of iron dextran so for mild to moderate anemia, we don't necessarily want to go to it first. We want to go to something with less of those side effects, like the oral therapy.
I can think of a psychiatric example as well for the second-generation antipsychotics, clozapine. Clozapine has a number of boxed warnings that other antipsychotics in the second generation do not have, including myocarditis, severe neutropenia, orthostatic hypotension, seizures. And it's not to say that clozapine is not an effective medication, but when we have a patient coming in for first episode psychosis or schizophrenia, clozapine may not be the first medication that we choose for that patient because of these more severe, disabling side effects.
2. Second, drugs with the potential for serious adverse reactions that may be prevented or reduced in severity by appropriate prescribing. For example, we want to avoid angiotensin-converting enzyme inhibitors [ACE] inhibitors in pregnant women during the second and third trimester because of potential birth defects. If we can use something else, that can be a good idea because we avoid that potential side effect.
Another example is liver function tests for valproic acid (Depakene). The boxed warning is for hepatotoxicity, especially in the first six months of treatments, and the warning actually suggests that liver function tests (LFTs) are taken at baseline and frequently, especially in those first six months.
3. Third, drugs with mandatory restrictions to ensure safe use. So, for example, prescribers must complete a certification program (iPledge) before prescribing isotretinoin, formerly Accutane. The reason they do that is because of the risk of teratogenicity and to minimize fetal exposure.
I can think again of clozapine in this example as a psychiatric example. We're all familiar with the fact that clozapine requires certification, training, and to be enrolled in the Risk Evaluation and Mitigation Strategy (REMs) program. We have to report the ANC, the absolute neutrophil count monthly and follow the guidelines for monitoring laboratory values regularly, and this is because the FDA has come in to say that we really need to do that close of monitoring because of the potential for adverse risks.
It's not a perfect process. The process by which a boxed warning is added to a drug is not necessarily systematic and may be affected by evidence, subjective choices, and even conflicts of interest.
Antidepressants and suicide risk
Broadly, this is defined as an increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants.
Antidepressants with increased suicide risk
Here we have a list of medications that have the boxed warning for increased suicide risk. What this overall shows is that the class of antidepressants has this boxed warning. That includes SSRI’s, SNRI’s atypical antidepressants, TCA’s and antipsychotics used for adjunctive treatment of depression.
|
|
Source: FDA. Suicidality in Children and Adolescents Being Treated With Antidepressant Medications | 2018 | |
Back story of antidepressants
Let's talk about the back story of antidepressants and where this boxed warning came from. In 1987, Prozac was the first selective serotonin reuptake inhibitor (SSRI) approved. In 1987, it was considered the wonder drug. It was different from TCAs because it didn't have as many side effects. It was still effective, and it didn't have that potentially lethal overdose risk that TCAs had.
Still in the 90s and the early to mid-90s, there was a question in some smaller studies as to whether antidepressants generally increased suicidal thinking, but none of them really came to the mainstream literature or evaluation of researchers. Fast forward to 2003, and this is when I would say the SSRI antidepressant, world of treatment for children/adolescents was turned upside down.
In 2003, Great Britain’s Medicines and Healthcare Products Regulatory Agency (MHRA), which is very similar to what the FDA is for the United States, reviewed clinical data on 1200 children treated with paroxetine for depression, social anxiety, and obsessive-compulsive disorder. This was data that paroxetine had gathered over many years prior. I'm not sure why the data all got released at once, but when it did, it really changed the landscape.
Soon after, in May 2003, Great Britain looked over this data and found that children with depression did not benefit from taking the drug. There was actually an increased risk of suicidal behavior among those treated with the drug compared with those receiving a placebo. The numbers were small, but they were significant. And then, Great Britain completely banned paroxetine for anyone under the age of 18. Of course, a few days later, the US took great notice to this, and they provided a public service announcement against the use of paroxetine for depressed youth until additional data could be examined.
And then as we end 2003, the UK decided to ban all use of antidepressants other than Prozac for the treatment of depression in children and adolescents. At this point, the US was still reviewing and we’ll talk about what they were reviewing and how they went over that.
US vs UK analyses
We can see that the UK took a pretty firm, maybe some would even say extreme, stance on antidepressants in children by banning them. But let's talk about the differences in the analysis between the US and UK and how the US got to the boxed warning.
One of the big differences between the FDA and the MHRA was that the FDA conducted an independent reclassification of suicidality. The FDA was concerned that the data did not use consistent measurements of suicidality across trials, so they ended up getting a group of 10 pediatric suicidologists organized by Columbia University led an independent and blind reclassification. When they were looking at the data from the studies of antidepressants, they classified suicide into five different categories, including suicide attempt preparatory actions towards imminent suicidal behavior, suicidal ideation, self-injury with intent unknown, and injury events. Essentially, they were doing a meta-analysis with reclassified data, and what they ended up finding was that all of the antidepressants increased the risks of suicidality among pediatric patients with depression.
Initially, the American Academy of Child and Adolescent Psychiatry (AACAP) announced that it did not support the issuance of black box warnings, stating that the data did not justify such a strong warning for antidepressants. However, soon after the FDA accepted the boxed warning, in a press release, AACAP said that it applauded the careful consideration the FDA has shown in issuing the new warning. Essentially, they changed their tune pretty quickly. In September 2004, the FDA panel reviewed the box warnings. A 23-member advisory panel and voted 15 to 8 in favor of black box warnings.
A few years later, in 2006, the FDA extended the warning to young adults aged up to 25 years, and this came from a meeting in December 2006 that was considered a contentious public session in which the advisory committee heard from psychiatric experts and from aggrieved family members who sometimes expressed outrage and anger at the FDA when they spoke of the death of a loved one who had taken antidepressants. In the end, the committee voted 6 to 2 in favor of extending the black box warning to include adults 18 to 24 years of age.
Then the next year in 2007, an expanded black box warning stated that depression itself was associated with an increased risk of suicide, so the FDA wanted to ensure that there was a clear understanding that antidepressants were not the only cause or precipitant to suicidality. Sometimes people with untreated depression can have suicidality.
History
Let's go into the back story of the back story and the studies that led to the banning of paroxetine in the UK. There were five studies in pediatrics conducted in the 1990s and 2000s for GlaxoSmithKline's paroxetine, also known as Seroxat in the UK and Paxil in the United States.
Three of the studies showed paroxetine no more effective than placebo. One study even found placebo to be more effective than paroxetine. There was clear data of increased suicidality in those taking the drug compared to placebo. It was even doubled. And one of the studies was infamously known as Study 329, and we'll talk about the controversy that came from that.
Why did GSK want to release these previously unpublished results to regulators in 2002? Well, it was four years after the first negative results appeared when the company hoped to win approval for the treatment of obsessive-compulsive disorder with paroxetine. To give you some context of how paroxetine was doing as a company in 2002, American physicians wrote 30.4 million prescriptions for paroxetine. The company made $2.5 billion from the United States and had total worldwide sales of $4.93 billion in the year 2002. Going into Study 329, this study was authored by the chair of psychiatry at Brown University in the United States. Later on, documents casted doubt on whether he actually analyzed the data himself and if he even wrote the final paper.
It later was found out that a ghost writer for a medical public relations company hired by GlaxoSmithKline, wrote the initial preparation of the study manuscript. In it, they made claims about the drug’s efficacy and had downplayed safety concerns. In July 2001, Study 329 was published in the Journal of the American Academy of Child Adolescent Psychiatry (JAACAP), which listed Keller, the psychiatrist from Brown and 21 other researchers as coauthors. The JAACAP article did include the negative results; however, it did not account for them in the conclusion. To the contrary, it concluded that paroxetine was “generally well tolerated and effective for major depression in adolescence.” They also said that it had remarkable “efficacy and safety in the treatment of adolescent depression.” Some even say that the company relied on the JAACAP article to promote paroxetine for off-label use in teenagers.
What ended up happening with this article? Well, from JAACAP's perspective, it never was retracted. The journal’s editors say the negative findings are included in the table, and therefore there are no grounds to withdraw the article [see the full PDF here].
This is not the same opinion of the British Medical Journal, who in September 2015 published a reanalysis of the study that included some of the negative results.
This controversy led to several lawsuits and strengthened calls for drug companies to disclose all their clinical research data. The MHRA even launched a criminal inquiry into GSK’s conduct but announced in 2008 that there would be no charges. In 2004, New York State Attorney Elliot Spitzer sued GSK for having withheld data. And in 2012, the United States Department of Justice fined the company $3 billion, including a sum for withholding data on paroxetine, unlawfully promoting it for the under 18s, and preparing a misleading article on Study 329.
What was the company's take? Their take was that they never withheld data, and they said that it was only when the data from its pediatric trials on paroxetine was analyzed together that they found an increased rate of suicidal thinking or attempted suicide was revealed. That may not be the exact picture because a memo was released from some top leadership of the company that described the release of paroxetine and the results from it as being “commercially unacceptable” and the company would need to “effectively manage the dissemination of these data in order to minimize any potential negative impact.”
Ultimately, I'll let you decide about how much the paroxetine knew about the negative side effects of its medication.
Breaking down the black box warning
Now that we’ve discussed the history leading up to the boxed warning, let's actually break down it down. This can be found on the FDA website for any drug that has the boxed warning for increased suicidal thinking, and we'll break it down into four different parts.
1. In the first part, they discuss that all ages can experience depression with worsening suicidal ideation. They pool data from short term trials that show that the drugs can increase the risk of suicidal thinking and behavior for those under the age of 25.
2. In the second part, they discussed that they used 24 short-term trials of nine antidepressants of 4400 pediatric patients. All drugs in the younger population showed increased risk of suicidality.
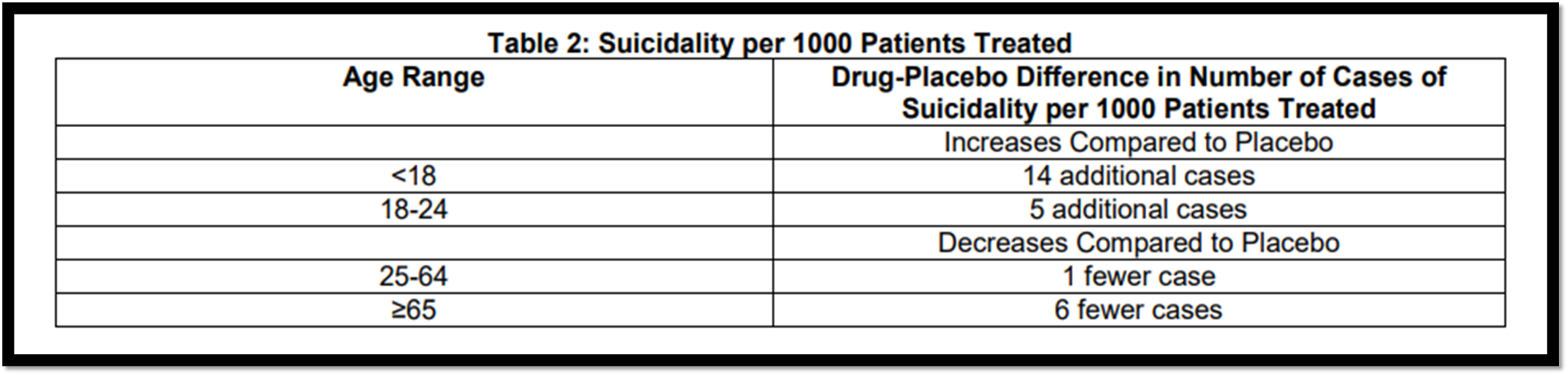
This is a table showing the suicidality per 1000 patients treated by age, and what you can see is that for under 18 years old, there were 14 additional cases per 1000 patients treated of suicidal thinking. For the 18 to 24, there were five additional cases, and then once you got to the adult range above 25, the rates of suicidality decreased compared to placebo, and even over 65 decreased even further.
3. There were NO completed SUICIDES in any of the pediatric trials. It is unknown whether the risk of suicidal ideation in pediatric patients extends to longer-term use because most of the studies were only a few months long. Overall, the average risk of suicidality for placebo was 2% and for medication was 4%. We must closely monitor symptoms when starting antidepressants especially in the beginning of treatment.
4. Lastly, if there are side effects, consider adjusting plan. Also, families and patients should be alerted to the importance of monitoring symptoms. That whole section is bolded. The takeaway message here is the importance of regularly assessing and reassessing the plan involving both the family and patient regularly.
Do antidepressants actually increase suicidality?
There's great information about this in The Carlat Child Psychiatry Report that goes into depth about this question. And I'm going to give an overview.
In the 1990s to 2004, antidepressant prescriptions were increasing. When the boxed warning passed from 2004 to 2009, antidepressant prescriptions were decreasing. And there were some studies that showed that there were increased suicides within United States for children/adolescents secondary to the prescriptions decreasing. However, this was not replicated in the UK where they also had lower antidepressant scripts without an increase in suicides.
1990-2004 – antidepressant prescriptions increased 2004-2009 – antidepressant prescriptions decreased 2009-present – antidepressant increasing, suicide rates increasing |
Additionally, the researchers connecting increased suicidality to decreased antidepressants, could have had some biases. Some of their studies utilized depression scales instead of suicide scales for measuring suicidality. Additionally, some of the studies only looked at one year of time, and the research really shows you need to look at trends over multiple years, not just one year, because it may go up or go down and not be caused by any specific factors.
Then in 2009 to present antidepressants were increasing, and suicide rates are increasing. Now, what does that mean? Well, it doesn't mean that antidepressants are causing suicidality necessarily, just as much as it doesn't mean that antidepressants don't create any risk for suicidality. We need to always tell the patient and the family about the risk for increased suicidal thinking for an antidepressant.
I want to remind you that any antidepressant that is used to treat children/adolescents has this boxed warning. This includes quetiapine, brexiprazole, aripiprazole, which are FDA approved as adjunctives for depression. Thus, they also have a boxed warning for increased suicidality in kids. I can't tell you why exactly, but olanzapine, even though it’s FDA approved for depression adjunctive does not have this warning. But if I were to use this in a child adolescent as an adjunctive, I would document that I talked to the family about this increased suicidal risk.
What's your role when prescribing antidepressants?
- DISCUSS the risks with your patient and family before prescribing
- Remind them of the likelihood
- Be understanding and compassionate about calling with questions
- Make clear what is something family should call you about vs an emergency
- With any prescription make sure you evaluate with family the risks and benefits
- Make sure you’re appropriately prescribing
- Consider the risks of not taking a prescription medication, too
The first one and the main one is that you need to discuss the risk with your patient and family before prescribing. This includes young children. You don't have to go into specifics, but I will say something like, “If you feel like hurting anybody, or yourself, you need to tell mommy or daddy or your teacher.” That's part of safety planning and that's something that you can document. You want to remind them of the likelihood. You want to be understanding and compassionate about calling with questions. You want to make clear what is something family should call you about versus an emergency. With any prescription, make sure you evaluate with family the risks and benefits. I also document that each time discuss the risks, benefits, alternatives, and side effects. You want to make sure you're appropriately prescribing, and you want to consider the risks of not taking a prescription, too.
What does the FDA suggest for antidepressant monitoring?
They suggest that once an antidepressant is started a clinician should monitor weekly for 4 weeks, then every other for 4 weeks, as 12 weeks and then as clinically indicated. Honestly, my clinic is booked out for at least 3 months for follow up appointments so this schedule would just not be possible for me. The big takeaway here is that when we are first initiating antidepressants, this is the time of greatest risk for side effects and increased suicidal thinking so we need to have closer monitoring at this vulnerable time.
FDA guidelines for antidepressant monitoring when first initiating a medication:
- Weekly for 4 weeks
- Every other week for 4 weeks
- At 12 weeks
- As clinically indicated beyond 12 weeks
Such monitoring would generally include at least weekly face-to-face contact with patients or their family members or caregivers during the first 4 weeks of treatment, then every other week visits for the next 4 weeks, then at 12 weeks, and as clinically indicated beyond 12 weeks. Additional contact by telephone may be appropriate between face-to-face visits.
Suicidal ideation
What happens when a patient has increased suicidal thoughts after starting an antidepressant? Really, there's not a perfect answer that I can give you, but I can tell you that it takes a multifactorial assessment and decision that should involve the patient, guardian, and provider. And overall, the big thing to take away is that context is really important to assess.
- Have there been recent stressors or changes that could be contributing to the increased suicidal thinking?
- Are there sleep changes?
- What's the medication history?
- Are they actually taking the medication?
- Are they taking it every few days, which could certainly lead to increased suicidal thinking?
- Are they taking it as prescribed?
- Are they taking half a pill, a full pill, two pills?
- And what does the patient/guardian think is the cause? I will directly ask the patient, “Do you think it's the antidepressant causing your thoughts?”
Answers will help guide you on what to do next in terms of lowering the medication, stopping the medication, or continuing or titrating the medication.
Conclusions
Antidepressants have the risk of increasing suicidal ideation. Untreated depression also has the risk of increasing suicidal ideation and suicide attempt completion. Antidepressants, fluoxetine, and escitalopram are FDA approved for depression in children in the US, and these are going to be the medications that are my first-line for children presenting with depression.
No matter what disorder you are treating primarily, it is recommended to always have a discussion with patient and family on the black box warning. And I want to leave you with a quote from Dr. David Healy of the University of Wales College of Medicine, who stated, “The issue isn't necessarily that these drugs should be banned for all children. The question is, ‘What is the safest way they can be used?’”
Thank you for joining this Carlat Webinar.
Earn CME for watching the webinars with a Webinar CME Subscription.
__________
The Carlat CME Institute is accredited by the ACCME to provide continuing medical education for physicians. Carlat CME Institute maintains responsibility for this program and its content. Carlat CME Institute designates this enduring material educational activity for a maximum of one-half (.5) AMA PRA Category 1 CreditsTM. Physicians or psychologists should claim credit commensurate only with the extent of their participation in the activity.



