
_-The-Breakthrough-Antipsychotic-That-Could-Change-Everything.webp?t=1729528747 "KarXT (Cobenfy)_ The Breakthrough Antipsychotic That Could Change Everything.jpg")
The Facts on Tardive Dyskinesia
We’re all aware that neuroleptics and other dopamine-receptor blocking agents (DRBAs) can cause a variety of movement disorders, often referred to as extrapyramidal syndromes (EPS).
Many EPS, such as acute dystonia, akathisia, or drug-induced parkinsonism, typically manifest within hours or days (sometimes a few weeks) after patients start neuroleptics, or after the dose is increased. Fortunately, these neuroleptic-induced movement disorders are reversible, and the symptoms resolve soon after the causative medication is discontinued.
This is not the case with tardive dyskinesia (TD), a fascinating disorder, or rather a group of drug-induced movement disorders with distinct clinical presentations. The main differences between TD and other drug-induced movement disorders are:
According to DSM-5, TD can develop after neuroleptics are used for at least a few months, or an even shorter period in older patients, and lasts “at least a few weeks.” TD can also result from sudden discontinuation or dose reduction of a neuroleptic, emerging within four to eight weeks of withdrawal and persisting for longer. When the symptoms last fewer than four to eight weeks after discontinuation of the offending drug, it’s called withdrawal-emergent dyskinesia (American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed.). Arlington, VA: American Psychiatric Publishing;2013).
Epidemiology
Up to 20% to 50% of patients treated with neuroleptics might develop TD, especially if they are treated with first generation, or “typical” antipsychotics. Second generation neuroleptics are associated with lower risk of TD (Correll CU et al, Am J Psychiatry 2004;161:414–425), although this has been contested (see TCPR, January 2013).
Middle-age and elderly patients have about a 10-fold higher risk of developing TD after one year of treatment compared to younger patients. Although both genders can develop TD, postmenopausal women might be at higher risk. Tardive dystonia is the only exception as it occurs more often in younger males.
Clinical Course and Types of TD
Tardive dyskinesia has an insidious onset with evolution into a full syndrome over days and weeks, and a waxing and waning course. The symptoms persist for years or decades in most patients, even after discontinuation of an offending drug. The severity of TD can range from mild movements that the patient might not be aware of, to a very severe, disabling disease.
Tardive dyskinesia can manifest as a variety of abnormal movements including stereotypy, dystonia, akathisia, tics, myoclonus, tremor, parkinsonism, or chorea (see box below) (Waln O & Jankovic J, Tremor Other Hyperkinet Mov 2013;3:1–11). Videos of these conditions can be found at http://1.usa.gov/1iWToMC.
Classic TD is the most common type of the disorder that manifests as repetitive oro-facial movements, or stereotypies, such as lip smacking, chewing, facial grimacing, tongue movements inside the mouth, and tongue popping out. Some patients with classic TD may also have stereotypic movements of the limbs or trunk such as foot tapping, piano-playing movements of fingers and toes, hands rubbing, trunk rocking, and swaying.
Tardive akathisia is a feeling of internal restlessness that is usually uncomfortable and can be disabling. These patients appear restless and fidgety. They often rock in a chair, cross-uncross legs when sitting, get up and pace on a spot, touch their face or scalp, or scratch themselves. Some can have repetitive moaning and grunting.
Tardive dystonia can be generalized or focal, usually affecting tongue and facial muscles, and it has very characteristic features. Patients with tardive dystonia typically have opisthotonic trunk arching, neck retrocollis, adduction and pronation of arms with extension of the elbows, and flexion of the wrists.
Tardive chorea in adult TD patients usually accompanies classic oro-facial dyskinesia. But in children, generalized chorea affecting mainly the trunk and limbs with no or minimal involvement of the oro-facial region, is a characteristic feature of withdrawal-emergent dyskinesia, a self-limiting condition typically resolving over several weeks.
Tardive tics are clinically indistinguishable from motor and phonic tics of Tourette syndrome, but much older age at onset in conjunction with the history of chronic exposure to a DRBA would point towards tardive tics.
Tardive tremor manifests as a high amplitude and low frequency postural, kinetic, and resting tremor with no other parkinsonian features such as bradykinesia, rigidity, or gait changes.
Tardive myoclonus presents as postural jerk-like movements usually affecting upper extremities.
Tardive parkinsonism can be diagnosed if parkinsonian features (ie, rest tremor, bradykinesia, rigidity, gait changes) persist for several months or years after discontinuation of a DRBA. Note that drug-induced parkinsonism typically resolves within a few weeks after stopping an offending drug. Both conditions should have normal dopamine transporter SPECT imaging (DaTscan), as opposed to Parkinson’s disease.
About one third of TD patients might have a combination of two or more tardive syndromes.
Etiology/Pathophysiology
Although neuroleptics are the most common cause of TD, antiemetics, vasoactive medications, and other psychotropic drugs, such as antidepressants, can also cause TD (See box below).
The pathophysiology of TD is still poorly understood (Waln O & Jankovic J, op.cit). One of the most popular theories about pathogenesis of TD is that chronic blockade of dopamine receptors in basal ganglia, particularly D2 and possibly D3, leads to the receptors’ upregulation (increasing in number) and supersensitivity to dopamine, producing a hyperkinetic movement disorder.
“Typical” antipsychotics bind to D2 receptors stronger and for a longer time than newer generation neuroleptics, and therefore they carry a higher risk of causing TD. “Atypical” neuroleptics, particularly clozapine (Clozaril) and quetiapine (Seroquel), have the lowest D2 receptor occupancy and therefore a lower risk of TD.
This receptor-supersensitivity theory makes sense if you recall that TD can manifest after sudden discontinuation of a neuroleptic, in which supersensitive dopamine receptors “crave the drug.” We also know that increasing the dose of a neuroleptic in patients who develop TD can temporarily improve the symptoms, possibly because the higher dose saturates the increased number of the receptors.
But wouldn’t you expect the receptors eventually to undergo downregulation, or decrease in number, after not being exposed to a DRBA for months? If so, TD symptoms should resolve, but this doesn’t happen in many cases. The neurodegenerative hypothesis and oxidative stress hypothesis of TD may explain why. According to these theories, neurons may be irreversibly damaged by neuroleptic-induced lipid peroxidation and free-radical formation, which causes persistent TD long after the discontinuation of the offending drug.
The most recent “maladaptive synaptic plasticity” hypothesis attempts to connect all previous theories and fill the gaps between them (Teo JT et al, Mov Disord 2012;27:1205–1215). According to this hypothesis, D2-receptor hypersensitivity and neuronal damage caused by increased oxidative stress can result in the formation of abnormal new connections between basal ganglia and brain cortex, leading to abnormal movements that persist even after the offending drug is removed.
The mechanism of TD caused by other, non-neuroleptic, medications is unclear. It is possible that increased levels of serotonin caused by SSRI and SNRI antidepressants can imitate an antidopaminergic effect on basal ganglia neurons similar to DRBAs, but this is not proven.
You’ve probably noticed from your own clinical experience that most patients taking antipsychotic medications for years do not develop TD, and the same medication regimen can cause different types and severity of TD. Does it mean there is genetic susceptibility for TD? Multiple gene candidates were recently found in association with TD, including genes coding for dopamine and serotonin receptors, antioxidative enzymes, and drug metabolism enzymes (Teo JT et al, op.cit). Therefore, although not a genetic disorder, TD might have a genetic predisposition.
Management of TD
Prevention of TD is paramount. Whenever you can, avoid DRBAs and choose alternative medications or “atypical” neuroleptics with lower potential to cause TD. If you decide to use DRBAs in your patient, frequently re-assess the need to continue treatment and watch for symptoms of TD at every visit.
Take action as soon as you notice signs of TD in your patient. The most important approach in treatment of TD is the removal of an offending drug as soon as possible. Remember, the longer patients with the symptoms of TD continue taking DRBAs, the lower their chances of recovery. But do not stop a DRBA abruptly; instead, taper it off slowly to avoid worsening of TD.
Although we know that increasing the dose of a DRBA can temporarily improve the symptoms of TD, this strategy should be avoided and used only in the most severe cases when immediate control of the disabling TD symptoms is required.
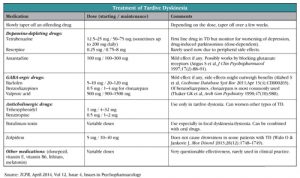
The dopamine-depleting drugs, reserpine (Serpalan) and tetrabenazine (Xenazine), are the most effective medications in any TD syndromes. Both medications prevent reuptake and recycling of the monoamines (including dopamine), thereby promoting their breakdown within the synaptic cleft. As a result, hypersensitive dopamine receptors receive reduced dopaminergic stimulation, thus improving hyperkinetic movements in the absence of dopamine-receptor blockade.
Tetrabenazine is currently considered a first-line treatment option in TD if the symptoms persist after discontinuation of a DRBA. Despite this, the use of tetrabenazine remains off label. So far, it is FDA-approved only for Huntington’s chorea, and is only available in the USA from a specialty pharmacy (see the manufacturer’s website, www.xenazineusa.com/HCP).
This medication is usually well tolerated; however it can cause or exacerbate depression, produce daytime somnolence, akathisia, and parkinsonism, especially in higher doses. Reserpine can cause the same side effects but also orthostatic hypotension and diarrhea; therefore it is used less often.
Other, less studied medications showed some symptomatic improvement in TD according to small studies and case reports. Among those medications are amantadine (Symmetrel), clonazepam (Klonopin) and other benzodiazepines, baclofen (Lioresal), valproic acid (Depakene), zolpidem (Ambien), donepezil (Aricept), lithium, antioxidants, zonisamide (Zonegran), vitamin B6, and melatonin. Anticholinergic medication like trihexyphenidyl (Artane) and benztropine can be effective in tardive dystonia but can exacerbate other types of TD.
Botulinum toxin injections are often used for treatment of focal dyskinesia and tardive dystonia. In the most severe and refractory cases of TD, surgical treatment such as pallidotomy or deep brain stimulation can be considered.
TCPR’s Verdict: TD can manifest as a variety of movement disorders and can be caused by medications other than first-generation antipsychotics. Early recognition of TD symptoms and discontinuation of the offending drug can improve prognosis in TD patients. Tetrabanazine is the first-line in treatment of any type of TD.
General PsychiatryMany EPS, such as acute dystonia, akathisia, or drug-induced parkinsonism, typically manifest within hours or days (sometimes a few weeks) after patients start neuroleptics, or after the dose is increased. Fortunately, these neuroleptic-induced movement disorders are reversible, and the symptoms resolve soon after the causative medication is discontinued.
This is not the case with tardive dyskinesia (TD), a fascinating disorder, or rather a group of drug-induced movement disorders with distinct clinical presentations. The main differences between TD and other drug-induced movement disorders are:
- the delayed onset of the symptoms, hence the name “tardive,” and
- persistence of the symptoms for months or years after a causative drug was discontinued.
According to DSM-5, TD can develop after neuroleptics are used for at least a few months, or an even shorter period in older patients, and lasts “at least a few weeks.” TD can also result from sudden discontinuation or dose reduction of a neuroleptic, emerging within four to eight weeks of withdrawal and persisting for longer. When the symptoms last fewer than four to eight weeks after discontinuation of the offending drug, it’s called withdrawal-emergent dyskinesia (American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed.). Arlington, VA: American Psychiatric Publishing;2013).
Epidemiology
Up to 20% to 50% of patients treated with neuroleptics might develop TD, especially if they are treated with first generation, or “typical” antipsychotics. Second generation neuroleptics are associated with lower risk of TD (Correll CU et al, Am J Psychiatry 2004;161:414–425), although this has been contested (see TCPR, January 2013).
Middle-age and elderly patients have about a 10-fold higher risk of developing TD after one year of treatment compared to younger patients. Although both genders can develop TD, postmenopausal women might be at higher risk. Tardive dystonia is the only exception as it occurs more often in younger males.
Clinical Course and Types of TD
Tardive dyskinesia has an insidious onset with evolution into a full syndrome over days and weeks, and a waxing and waning course. The symptoms persist for years or decades in most patients, even after discontinuation of an offending drug. The severity of TD can range from mild movements that the patient might not be aware of, to a very severe, disabling disease.
Tardive dyskinesia can manifest as a variety of abnormal movements including stereotypy, dystonia, akathisia, tics, myoclonus, tremor, parkinsonism, or chorea (see box below) (Waln O & Jankovic J, Tremor Other Hyperkinet Mov 2013;3:1–11). Videos of these conditions can be found at http://1.usa.gov/1iWToMC.
Classic TD is the most common type of the disorder that manifests as repetitive oro-facial movements, or stereotypies, such as lip smacking, chewing, facial grimacing, tongue movements inside the mouth, and tongue popping out. Some patients with classic TD may also have stereotypic movements of the limbs or trunk such as foot tapping, piano-playing movements of fingers and toes, hands rubbing, trunk rocking, and swaying.
Tardive akathisia is a feeling of internal restlessness that is usually uncomfortable and can be disabling. These patients appear restless and fidgety. They often rock in a chair, cross-uncross legs when sitting, get up and pace on a spot, touch their face or scalp, or scratch themselves. Some can have repetitive moaning and grunting.
Tardive dystonia can be generalized or focal, usually affecting tongue and facial muscles, and it has very characteristic features. Patients with tardive dystonia typically have opisthotonic trunk arching, neck retrocollis, adduction and pronation of arms with extension of the elbows, and flexion of the wrists.
Tardive chorea in adult TD patients usually accompanies classic oro-facial dyskinesia. But in children, generalized chorea affecting mainly the trunk and limbs with no or minimal involvement of the oro-facial region, is a characteristic feature of withdrawal-emergent dyskinesia, a self-limiting condition typically resolving over several weeks.
Tardive tics are clinically indistinguishable from motor and phonic tics of Tourette syndrome, but much older age at onset in conjunction with the history of chronic exposure to a DRBA would point towards tardive tics.
Tardive tremor manifests as a high amplitude and low frequency postural, kinetic, and resting tremor with no other parkinsonian features such as bradykinesia, rigidity, or gait changes.
Tardive myoclonus presents as postural jerk-like movements usually affecting upper extremities.
Tardive parkinsonism can be diagnosed if parkinsonian features (ie, rest tremor, bradykinesia, rigidity, gait changes) persist for several months or years after discontinuation of a DRBA. Note that drug-induced parkinsonism typically resolves within a few weeks after stopping an offending drug. Both conditions should have normal dopamine transporter SPECT imaging (DaTscan), as opposed to Parkinson’s disease.
About one third of TD patients might have a combination of two or more tardive syndromes.
Neuroleptic-Induced Movement Disorders
Acute/Subacute (hours–weeks)
Acute dystonic reaction
Acute akathisia
Drug-induced parkinsonism
Neuroleptic malignant syndrome
(can also manifest after chronic exposure to neuroleptics)
Acute akathisia
Drug-induced parkinsonism
Neuroleptic malignant syndrome
(can also manifest after chronic exposure to neuroleptics)
Tardive (months–years)
Classic tardive dyskinesia
Tardive stereotypy
Tardive dystonia
Tardive akathisia
Tardive tics (tardive tourettism)
Tardive tremor
Tardive myoclonus
Tardive chorea
Tardive parkinsonism
Withdrawal emergent syndrome
Tardive stereotypy
Tardive dystonia
Tardive akathisia
Tardive tics (tardive tourettism)
Tardive tremor
Tardive myoclonus
Tardive chorea
Tardive parkinsonism
Withdrawal emergent syndrome
Etiology/Pathophysiology
Although neuroleptics are the most common cause of TD, antiemetics, vasoactive medications, and other psychotropic drugs, such as antidepressants, can also cause TD (See box below).
The pathophysiology of TD is still poorly understood (Waln O & Jankovic J, op.cit). One of the most popular theories about pathogenesis of TD is that chronic blockade of dopamine receptors in basal ganglia, particularly D2 and possibly D3, leads to the receptors’ upregulation (increasing in number) and supersensitivity to dopamine, producing a hyperkinetic movement disorder.
“Typical” antipsychotics bind to D2 receptors stronger and for a longer time than newer generation neuroleptics, and therefore they carry a higher risk of causing TD. “Atypical” neuroleptics, particularly clozapine (Clozaril) and quetiapine (Seroquel), have the lowest D2 receptor occupancy and therefore a lower risk of TD.
This receptor-supersensitivity theory makes sense if you recall that TD can manifest after sudden discontinuation of a neuroleptic, in which supersensitive dopamine receptors “crave the drug.” We also know that increasing the dose of a neuroleptic in patients who develop TD can temporarily improve the symptoms, possibly because the higher dose saturates the increased number of the receptors.
But wouldn’t you expect the receptors eventually to undergo downregulation, or decrease in number, after not being exposed to a DRBA for months? If so, TD symptoms should resolve, but this doesn’t happen in many cases. The neurodegenerative hypothesis and oxidative stress hypothesis of TD may explain why. According to these theories, neurons may be irreversibly damaged by neuroleptic-induced lipid peroxidation and free-radical formation, which causes persistent TD long after the discontinuation of the offending drug.
The most recent “maladaptive synaptic plasticity” hypothesis attempts to connect all previous theories and fill the gaps between them (Teo JT et al, Mov Disord 2012;27:1205–1215). According to this hypothesis, D2-receptor hypersensitivity and neuronal damage caused by increased oxidative stress can result in the formation of abnormal new connections between basal ganglia and brain cortex, leading to abnormal movements that persist even after the offending drug is removed.
The mechanism of TD caused by other, non-neuroleptic, medications is unclear. It is possible that increased levels of serotonin caused by SSRI and SNRI antidepressants can imitate an antidopaminergic effect on basal ganglia neurons similar to DRBAs, but this is not proven.
You’ve probably noticed from your own clinical experience that most patients taking antipsychotic medications for years do not develop TD, and the same medication regimen can cause different types and severity of TD. Does it mean there is genetic susceptibility for TD? Multiple gene candidates were recently found in association with TD, including genes coding for dopamine and serotonin receptors, antioxidative enzymes, and drug metabolism enzymes (Teo JT et al, op.cit). Therefore, although not a genetic disorder, TD might have a genetic predisposition.
Medications Associated with Tardive Dyskinesia
Antipsychotics
1st generation: haloperidol, loxapine, pimozide, chlorpromazine, thioridazine, trifluoperazine, prochlorperazine, perphenazine, fluphenazine, thiothixene
2nd generation: ziprasidone, iloperidone, asenapine, clozapine, quetiapine, risperidone, paliperidone, olanzapine, amisulpride, aripiprazole
2nd generation: ziprasidone, iloperidone, asenapine, clozapine, quetiapine, risperidone, paliperidone, olanzapine, amisulpride, aripiprazole
Antiemetics
droperidol, metoclopramide, chlorpromazine, prochlorperazine
Antidepressants
SSRI/SNRI: duloxetine, citalopram
Tricyclic: amoxapine
Tricyclic: amoxapine
Lithium
Calcium channel blockers
flunarizine, cinnarizine
Management of TD
Prevention of TD is paramount. Whenever you can, avoid DRBAs and choose alternative medications or “atypical” neuroleptics with lower potential to cause TD. If you decide to use DRBAs in your patient, frequently re-assess the need to continue treatment and watch for symptoms of TD at every visit.
Take action as soon as you notice signs of TD in your patient. The most important approach in treatment of TD is the removal of an offending drug as soon as possible. Remember, the longer patients with the symptoms of TD continue taking DRBAs, the lower their chances of recovery. But do not stop a DRBA abruptly; instead, taper it off slowly to avoid worsening of TD.
Although we know that increasing the dose of a DRBA can temporarily improve the symptoms of TD, this strategy should be avoided and used only in the most severe cases when immediate control of the disabling TD symptoms is required.
The dopamine-depleting drugs, reserpine (Serpalan) and tetrabenazine (Xenazine), are the most effective medications in any TD syndromes. Both medications prevent reuptake and recycling of the monoamines (including dopamine), thereby promoting their breakdown within the synaptic cleft. As a result, hypersensitive dopamine receptors receive reduced dopaminergic stimulation, thus improving hyperkinetic movements in the absence of dopamine-receptor blockade.
Tetrabenazine is currently considered a first-line treatment option in TD if the symptoms persist after discontinuation of a DRBA. Despite this, the use of tetrabenazine remains off label. So far, it is FDA-approved only for Huntington’s chorea, and is only available in the USA from a specialty pharmacy (see the manufacturer’s website, www.xenazineusa.com/HCP).
This medication is usually well tolerated; however it can cause or exacerbate depression, produce daytime somnolence, akathisia, and parkinsonism, especially in higher doses. Reserpine can cause the same side effects but also orthostatic hypotension and diarrhea; therefore it is used less often.
Other, less studied medications showed some symptomatic improvement in TD according to small studies and case reports. Among those medications are amantadine (Symmetrel), clonazepam (Klonopin) and other benzodiazepines, baclofen (Lioresal), valproic acid (Depakene), zolpidem (Ambien), donepezil (Aricept), lithium, antioxidants, zonisamide (Zonegran), vitamin B6, and melatonin. Anticholinergic medication like trihexyphenidyl (Artane) and benztropine can be effective in tardive dystonia but can exacerbate other types of TD.
Botulinum toxin injections are often used for treatment of focal dyskinesia and tardive dystonia. In the most severe and refractory cases of TD, surgical treatment such as pallidotomy or deep brain stimulation can be considered.
TCPR’s Verdict: TD can manifest as a variety of movement disorders and can be caused by medications other than first-generation antipsychotics. Early recognition of TD symptoms and discontinuation of the offending drug can improve prognosis in TD patients. Tetrabanazine is the first-line in treatment of any type of TD.


Recommended


