
_-The-Breakthrough-Antipsychotic-That-Could-Change-Everything.webp?t=1729528747 "KarXT (Cobenfy)_ The Breakthrough Antipsychotic That Could Change Everything.jpg")
Glutamate’s Role in Schizophrenia
When a patient or caregiver asks you about the biology of schizophrenia or to explain how antipsychotics work, your answer most likely involves some discussion of dopamine. After all, all existing antipsychotic drugs are dopamine blockers, and agents that promote dopamine release, such as amphetamines, can cause psychotic symptoms.
However, there’s likely to be far more to the story. Current antipsychotic medications typically reduce positive symptoms—such as hallucinations and delusions—but have very little effect on negative symptoms—such as social withdrawal and blunted affect—or the cognitive symptoms that are also a hallmark of schizophrenia (see Expert Q&A in this issue). As a result, much effort has been directed at other mechanisms underlying this complex disorder.
Glutamate: The Highs and Lows
There’s a rich history of research suggesting that glutamate may be involved in the pathogenesis of schizophrenia, with an increasing amount of attention being brought to glutamate during the past decade or so. First, researchers have identified excess glutamate in the cortex of patients with schizophrenia (Moghaddam B et al, J Neurosci 1997;17:2921–2927), and antipsychotic treatment correlates with a decrease in cortical glutamate (de la Fuente-Sandoval C et al, JAMA Psychiatry 2013;70(10):1057–1066).
Furthermore, genetic studies have linked genes involved in glutamate signaling to schizophrenia. Finally, and perhaps most convincingly, drugs that block the NMDA type of glutamate receptor (see box below) cause psychotic symptoms (see box below for a review of receptor pharmacology). In fact, phencyclidine (PCP) and ketamine are well known to cause not only positive symptoms of psychosis, but also the negative and cognitive symptoms, as well (Krystal JH et al, Arch Gen Psychiatry 1994;51:199–214).
To simplify, two key findings in schizophrenia are: (1) increased glutamate in the cortex, and (2) decreased function of NMDA receptors. One line of thinking goes like this: when NMDA receptors on inhibitory interneurons don’t work properly, there’s less inhibition (ie, there’s “disinhibition”) of downstream neurons and, consequently, more cortical glutamate release (Olney JW and Farber NB, Arch Gen Psychiatry 1995;52(12):998–1007).
Dysfunction in the glutamate system may also explain disruptions in dopamine: NMDA dysfunction may increase dopamine output—via deficient inhibitory interneurons, as mentioned previously—to the striatum (underlying positive symptoms of psychosis) while indirectly reducing dopamine output to the cortex via the mesocortical pathway (underlying negative symptoms and cognitive impairment) (Lewis DA and Gonzalez-Burgos G, Nat Med 2006;12(9):1016–1022).
Glutamate-Based Antipsychotics?
Despite the evidence in support of glutamate, why has this amino acid neurotransmitter received so much less attention than its monoamine cousin, dopamine? Why is there no glutamatergic antipsychotic on the market? One reason may be that glutamate is the most abundant neurotransmitter in the human brain, so any drug that affects glutamate signaling is likely to have widespread effects on multiple cognitive functions, not to mention alter the risk of seizure and excitotoxicity. Glutamate, to be sure, is far from an “easy target.”
Nevertheless, certain isolated abnormalities—such as NMDA receptor dysfunction—provide a more focused target for drug design. And given the complexity of the NMDA receptor, there are a wide range of therapeutic targets for pharmacological manipulation, all of which have received at least some attention in clinical research.
Some experimental glutamatergic antipsychotics focus on enhancing and augmenting the “co-transmitter” of glutamate, namely glycine. Any intervention that increases the quantity or duration of glycine (or a related amino acid, like D-serine) in the synaptic cleft could, theoretically, stimulate NMDA function.
Other efforts have focused on the metabotropic glutamate (mGlu) eceptors. These have not yet been mentioned here, but there are eight different mGlu receptors (mGlu1–mGlu8), of which two subtypes, mGlu2/3 and mGlu5, have been implicated in schizophrenia. Specifically, activation of the mGlu2/3 subtype can mitigate the cortical glutamate release found with NMDA blockade, and it can inhibit the dopamine release induced by PCP (Ellenbroek BA, Neuropharmacology 2012;62:1371–1380).
Glutamate in Clinical Trials
Currently available antipsychotics only induce full remission in about one-third of patients (Haro JM et al, Br J Psychiatry 2011;199:194–201), so it should be no surprise that drug companies have jumped on the glutamate bandwagon in a very big way. Almost a dozen glutamatergic compounds have been studied in clinical trials. Despite the enthusiasm, however, results have been rather equivocal.
Drugs enhancing glycine. Augmenting glutamate activity with one of its co-transmitters (glycine or D-serine) seems like a reasonable way to potentiate NMDA function. Small trials of these amino acids, as well as other related compounds like D-cycloserine, given together with antipsychotic medication, have shown some promise in reducing negative symptoms and enhancing some aspects of cognitive function (see, for instance, Lane HY et al, Arch Gen Psychiatry 2005;62:1196–1204).
These compounds are problematic, however, in that they penetrate the blood-brain barrier poorly and have a short half-life. Thus, several more recent clinical trials have focused on inhibiting the reuptake of glycine by glycine transporters. These “GlyT1 inhibitors” include the compound bitopertin, developed by Roche Pharmaceuticals. Data from phase II clinical trials showed that bitopertin (10 or 30 mg/day) led to significant improvement in negative symptoms relative to placebo. So Roche launched an ambitious phase III project involving six separate clinical trials enrolling 3,600 subjects. In January 2014 the results of two trials were announced. Unfortunately, bitopertin showed no improvement in negative symptoms when added to antipsychotics. The remaining trials are still under way.
Drugs activating mGlu receptors. Multiple lines of evidence suggest that agonists of the mGlu receptor subtypes 2 and 3 (ie, “mGlu2/3 agonists”) inhibit the excessive release of cortical glutamate seen when NMDA receptors are blocked. It’s not clear whether this is because mGlu2/3 receptors serve as “negative feedback” autoreceptors to inhibit further glutamate release, or for another reason.
A few years ago, Eli Lilly found positive results in a phase II clinical trial of their mGlu2/3 compound (pomeglumetad methionil, also known as LY2140023); specifically, subjects taking LY2140023 experienced reductions in their PANSS (Positive And Negative Symptom Scale) scores for positive symptoms (-6.4 points relative to placebo) and negative symptoms (-4.7 points). These reductions were comparable in magnitude to those seen with olanzapine (Zyprexa), but without the risk of extrapyramidal symptoms or weight gain (Patil ST et al, Nat Med 2007;13(9):1102–1107). Unfortunately, in a second phase II trial, neither LY2140023 nor its active comparator, olanzapine, performed any better than placebo (Kinon BJ et al, J Clin Psychopharmacol 2011;31(3):349–355). As a result, Lilly halted its phase III trial in August 2012. We probably won’t be hearing anything more about this drug.
Nevertheless, a few other mGlu2/3 agonists are in development, including one compound (ADX71149, Janssen and Addex Pharmaceuticals) that acts as a “positive allosteric modulator” (PAM) of the mGlu2 receptor. PAMs may represent a novel new pharmacologic strategy. They’re neither agonists nor antagonists; rather, they bind to the receptor and make it more active, but only in the presence of the endogenous ligand (in this case, glutamate). Thus, they augment and enhance normal signaling rather than exerting a chronic effect.
Other glutamatergic agents. In addition to glycine transport inhibitors and mGlu2/3 agonists, other drugs that target specific subunits of the NMDA receptor and other mGlu receptor subtypes are under investigation. Still other research efforts are focusing on activators of the AMPA-type glutamate receptor (the so-called “AMPA-kines”).
While it’s counterintuitive that activating glutamate receptors might help treat a disease that’s thought to be due to excess glutamate in the first place, it’s quite possible that targeted activation of specific glutamate receptors (such as the AMPA type) might be useful. Indeed, at least two AMPA-kines have been studied for enhancement of cognition in schizophrenia, albeit both with negative results, and others are being evaluated as cognitive enhancers for other disorders (Lynch G & Gail CM, Trends Neurosci 2006;29(10):554–562).
No drugs acting on the glutamate system have yet been approved for the treatment of schizophrenia—and the most promising candidates, such as bitopertin and LY2140023, have failed late-stage trials. Nevertheless, there appears to be a great deal of excitement and momentum around this novel approach. Some have argued that patients who fail to improve on dopaminergic antipsychotics may suffer from more of a glutamate signaling deficiency. Perhaps someday, these patients can be identified via brain scanning, genotype analysis, or other biomarkers (Egerton A et al, Neuropsychopharmacology 2012;37:2515–2521).
TCPR’s Verdict: Glutamatergic antipsychotics are not yet available, but the day when glutamate becomes part of your schizophrenia vocabulary may be right around the corner. Understanding the terminology and the basic biology, and deciphering clinical trial results when available, will be important in determining whether glutamate-based agents have a place in your practice.
No need to dust off those old medical school textbooks: Here's a quick review of receptor pharmacology.
Glutamate receptors take two forms: ion channels that are "gated" by glutamate (the so-called ionotropic receptors); and seven-transmembrane-domain proteins with intracellular actions (the so-called metabotropic receptors, sometimes abbreviated "mGlu").
There are, in turn, three main types of ionotropic receptors: the NMDA subtype, the AMPA subtype, and the kainate subtypes. The NMDA receptor has received much attention due to its role in long-term potentiation (LTP) in the hippocampus, which is thought to underlie learning and memory, and as a target for new therapeutics for depression such as intravenous ketamine (see interview, TCPR Nov 2013).
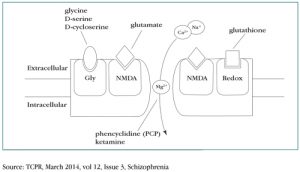
The NMDA receptor is also very complex (see figure). It's activated only when (a) glutamate binds, (b) the cell is depolarized, and (c) a co-ligand such as glycine or serine binds, as well. Thus, it's sometimes called a "coincidence detector." The NMDA receptor can be blocked by PCP and ketamine, each of which has its own binding site in the channel itself, and by magnesium ions, which are ejected when the cell is depolarized.
General PsychiatryHowever, there’s likely to be far more to the story. Current antipsychotic medications typically reduce positive symptoms—such as hallucinations and delusions—but have very little effect on negative symptoms—such as social withdrawal and blunted affect—or the cognitive symptoms that are also a hallmark of schizophrenia (see Expert Q&A in this issue). As a result, much effort has been directed at other mechanisms underlying this complex disorder.
Glutamate: The Highs and Lows
There’s a rich history of research suggesting that glutamate may be involved in the pathogenesis of schizophrenia, with an increasing amount of attention being brought to glutamate during the past decade or so. First, researchers have identified excess glutamate in the cortex of patients with schizophrenia (Moghaddam B et al, J Neurosci 1997;17:2921–2927), and antipsychotic treatment correlates with a decrease in cortical glutamate (de la Fuente-Sandoval C et al, JAMA Psychiatry 2013;70(10):1057–1066).
Furthermore, genetic studies have linked genes involved in glutamate signaling to schizophrenia. Finally, and perhaps most convincingly, drugs that block the NMDA type of glutamate receptor (see box below) cause psychotic symptoms (see box below for a review of receptor pharmacology). In fact, phencyclidine (PCP) and ketamine are well known to cause not only positive symptoms of psychosis, but also the negative and cognitive symptoms, as well (Krystal JH et al, Arch Gen Psychiatry 1994;51:199–214).
To simplify, two key findings in schizophrenia are: (1) increased glutamate in the cortex, and (2) decreased function of NMDA receptors. One line of thinking goes like this: when NMDA receptors on inhibitory interneurons don’t work properly, there’s less inhibition (ie, there’s “disinhibition”) of downstream neurons and, consequently, more cortical glutamate release (Olney JW and Farber NB, Arch Gen Psychiatry 1995;52(12):998–1007).
Dysfunction in the glutamate system may also explain disruptions in dopamine: NMDA dysfunction may increase dopamine output—via deficient inhibitory interneurons, as mentioned previously—to the striatum (underlying positive symptoms of psychosis) while indirectly reducing dopamine output to the cortex via the mesocortical pathway (underlying negative symptoms and cognitive impairment) (Lewis DA and Gonzalez-Burgos G, Nat Med 2006;12(9):1016–1022).
Glutamate-Based Antipsychotics?
Despite the evidence in support of glutamate, why has this amino acid neurotransmitter received so much less attention than its monoamine cousin, dopamine? Why is there no glutamatergic antipsychotic on the market? One reason may be that glutamate is the most abundant neurotransmitter in the human brain, so any drug that affects glutamate signaling is likely to have widespread effects on multiple cognitive functions, not to mention alter the risk of seizure and excitotoxicity. Glutamate, to be sure, is far from an “easy target.”
Nevertheless, certain isolated abnormalities—such as NMDA receptor dysfunction—provide a more focused target for drug design. And given the complexity of the NMDA receptor, there are a wide range of therapeutic targets for pharmacological manipulation, all of which have received at least some attention in clinical research.
Some experimental glutamatergic antipsychotics focus on enhancing and augmenting the “co-transmitter” of glutamate, namely glycine. Any intervention that increases the quantity or duration of glycine (or a related amino acid, like D-serine) in the synaptic cleft could, theoretically, stimulate NMDA function.
Other efforts have focused on the metabotropic glutamate (mGlu) eceptors. These have not yet been mentioned here, but there are eight different mGlu receptors (mGlu1–mGlu8), of which two subtypes, mGlu2/3 and mGlu5, have been implicated in schizophrenia. Specifically, activation of the mGlu2/3 subtype can mitigate the cortical glutamate release found with NMDA blockade, and it can inhibit the dopamine release induced by PCP (Ellenbroek BA, Neuropharmacology 2012;62:1371–1380).
Glutamate in Clinical Trials
Currently available antipsychotics only induce full remission in about one-third of patients (Haro JM et al, Br J Psychiatry 2011;199:194–201), so it should be no surprise that drug companies have jumped on the glutamate bandwagon in a very big way. Almost a dozen glutamatergic compounds have been studied in clinical trials. Despite the enthusiasm, however, results have been rather equivocal.
Drugs enhancing glycine. Augmenting glutamate activity with one of its co-transmitters (glycine or D-serine) seems like a reasonable way to potentiate NMDA function. Small trials of these amino acids, as well as other related compounds like D-cycloserine, given together with antipsychotic medication, have shown some promise in reducing negative symptoms and enhancing some aspects of cognitive function (see, for instance, Lane HY et al, Arch Gen Psychiatry 2005;62:1196–1204).
These compounds are problematic, however, in that they penetrate the blood-brain barrier poorly and have a short half-life. Thus, several more recent clinical trials have focused on inhibiting the reuptake of glycine by glycine transporters. These “GlyT1 inhibitors” include the compound bitopertin, developed by Roche Pharmaceuticals. Data from phase II clinical trials showed that bitopertin (10 or 30 mg/day) led to significant improvement in negative symptoms relative to placebo. So Roche launched an ambitious phase III project involving six separate clinical trials enrolling 3,600 subjects. In January 2014 the results of two trials were announced. Unfortunately, bitopertin showed no improvement in negative symptoms when added to antipsychotics. The remaining trials are still under way.
Drugs activating mGlu receptors. Multiple lines of evidence suggest that agonists of the mGlu receptor subtypes 2 and 3 (ie, “mGlu2/3 agonists”) inhibit the excessive release of cortical glutamate seen when NMDA receptors are blocked. It’s not clear whether this is because mGlu2/3 receptors serve as “negative feedback” autoreceptors to inhibit further glutamate release, or for another reason.
A few years ago, Eli Lilly found positive results in a phase II clinical trial of their mGlu2/3 compound (pomeglumetad methionil, also known as LY2140023); specifically, subjects taking LY2140023 experienced reductions in their PANSS (Positive And Negative Symptom Scale) scores for positive symptoms (-6.4 points relative to placebo) and negative symptoms (-4.7 points). These reductions were comparable in magnitude to those seen with olanzapine (Zyprexa), but without the risk of extrapyramidal symptoms or weight gain (Patil ST et al, Nat Med 2007;13(9):1102–1107). Unfortunately, in a second phase II trial, neither LY2140023 nor its active comparator, olanzapine, performed any better than placebo (Kinon BJ et al, J Clin Psychopharmacol 2011;31(3):349–355). As a result, Lilly halted its phase III trial in August 2012. We probably won’t be hearing anything more about this drug.
Nevertheless, a few other mGlu2/3 agonists are in development, including one compound (ADX71149, Janssen and Addex Pharmaceuticals) that acts as a “positive allosteric modulator” (PAM) of the mGlu2 receptor. PAMs may represent a novel new pharmacologic strategy. They’re neither agonists nor antagonists; rather, they bind to the receptor and make it more active, but only in the presence of the endogenous ligand (in this case, glutamate). Thus, they augment and enhance normal signaling rather than exerting a chronic effect.
Other glutamatergic agents. In addition to glycine transport inhibitors and mGlu2/3 agonists, other drugs that target specific subunits of the NMDA receptor and other mGlu receptor subtypes are under investigation. Still other research efforts are focusing on activators of the AMPA-type glutamate receptor (the so-called “AMPA-kines”).
While it’s counterintuitive that activating glutamate receptors might help treat a disease that’s thought to be due to excess glutamate in the first place, it’s quite possible that targeted activation of specific glutamate receptors (such as the AMPA type) might be useful. Indeed, at least two AMPA-kines have been studied for enhancement of cognition in schizophrenia, albeit both with negative results, and others are being evaluated as cognitive enhancers for other disorders (Lynch G & Gail CM, Trends Neurosci 2006;29(10):554–562).
No drugs acting on the glutamate system have yet been approved for the treatment of schizophrenia—and the most promising candidates, such as bitopertin and LY2140023, have failed late-stage trials. Nevertheless, there appears to be a great deal of excitement and momentum around this novel approach. Some have argued that patients who fail to improve on dopaminergic antipsychotics may suffer from more of a glutamate signaling deficiency. Perhaps someday, these patients can be identified via brain scanning, genotype analysis, or other biomarkers (Egerton A et al, Neuropsychopharmacology 2012;37:2515–2521).
TCPR’s Verdict: Glutamatergic antipsychotics are not yet available, but the day when glutamate becomes part of your schizophrenia vocabulary may be right around the corner. Understanding the terminology and the basic biology, and deciphering clinical trial results when available, will be important in determining whether glutamate-based agents have a place in your practice.
The Wide, Wide World Of Glutamate Receptors
No need to dust off those old medical school textbooks: Here's a quick review of receptor pharmacology.
Glutamate receptors take two forms: ion channels that are "gated" by glutamate (the so-called ionotropic receptors); and seven-transmembrane-domain proteins with intracellular actions (the so-called metabotropic receptors, sometimes abbreviated "mGlu").
There are, in turn, three main types of ionotropic receptors: the NMDA subtype, the AMPA subtype, and the kainate subtypes. The NMDA receptor has received much attention due to its role in long-term potentiation (LTP) in the hippocampus, which is thought to underlie learning and memory, and as a target for new therapeutics for depression such as intravenous ketamine (see interview, TCPR Nov 2013).
The NMDA receptor is also very complex (see figure). It's activated only when (a) glutamate binds, (b) the cell is depolarized, and (c) a co-ligand such as glycine or serine binds, as well. Thus, it's sometimes called a "coincidence detector." The NMDA receptor can be blocked by PCP and ketamine, each of which has its own binding site in the channel itself, and by magnesium ions, which are ejected when the cell is depolarized.


Recommended


