
_-The-Breakthrough-Antipsychotic-That-Could-Change-Everything.webp?t=1729528747 "KarXT (Cobenfy)_ The Breakthrough Antipsychotic That Could Change Everything.jpg")
EXPERT Q&A
Effects of Drug Interactions
 Neil Sandson, MD.
Neil Sandson, MD. Clinical Associate Professor, Department of Psychiatry at University of Maryland School of Medicine
Dr. Sandson has disclosed that he has no relevant financial or other interests in any commercial companies pertaining to this educational activity.
TCPR: We’re seeing a lot of computer alerts for drug interactions. Which ones should we pay attention to?
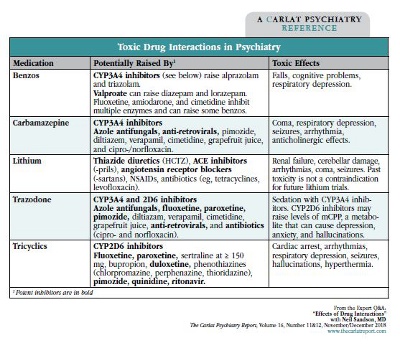
Dr. Sandson: The two issues to pay attention to are toxicity and loss of efficacy. Toxicity is the bigger concern, particularly if the drug has a “narrow therapeutic index,” where modest differences in the level can have dangerous effects. Lithium, tricyclics, carbamazepine, and to a lesser extent valproate are some psychiatric examples where high levels can cause problems (see “Toxic Drug Interactions in Psychiatry”). Drug interactions with inhibitors can cause high levels by blocking metabolism by P450 enzymes in the liver. The potency of the inhibitor also matters here. An inhibitor that raises a drug by only 20% is probably not going to cause much concern.
TCPR: What are some potent inhibitors we use?
Dr. Sandson: Fluoxetine (Prozac), fluvoxamine (Luvox), paroxetine (Paxil), and duloxetine (Cymbalta). Fluoxetine and fluvoxamine are potent and hit a broad array of P450 enzymes, while paroxetine’s effects are fairly specific for CYP2D6. Sertraline (Zoloft) is also an inhibitor at CYP2D6, but it doesn’t become potent until you reach the higher dosages, like 150 mg and above.
TCPR: What about inducers?
Dr. Sandson: Inducers do the opposite. They lower drug levels, and potent ones can drop the affected drugs out of the therapeutic range. The inducers often tend to be among the anticonvulsants, and the most potent one we use in psychiatry is carbamazepine (Equetro, Tegretol). Induction is usually not as dangerous as inhibition, but there are exceptions.
TCPR: When is induction a serious problem?
Dr. Sandson: The main examples are oral contraceptives, warfarin, and immunosuppressants (cyclosporins, sirolimus, tacrolimus), all of which are significantly induced by carbamazepine at CYP3A4. Carbamazepine can render birth control ineffective. Lowering immunosuppressants can lead to organ rejection in people who’ve had a transplant, and lowering warfarin can result in ischemic events through coagulation. High levels of warfarin are also dangerous, causing bleeding and hemorrhage, and CYP2C9 inhibitors will raise it.
TCPR: What about other anticonvulsants? Can they induce 3A4 like carbamazepine does?
Dr. Sandson: Other psych meds that induce 3A4 include St. John’s wort, modafinil (Provigil), armodafinil (Nuvigil), topiramate (Topamax), oxcarbazepine (Trileptal), and barbiturates. Several of these are less potent inducers, but you’d still want to avoid them when the risks of induction are serious. The actual interaction may vary by person, as well as by dose. Topiramate and oxcarbazepine’s induction gets more potent as the dosage is increased.
TCPR: Does this 3A4 interaction affect all forms of birth control?
Dr. Sandson: Estrogen and progesterone-based contraceptives will be affected by 3A4 induction, which includes most of them. Ethinyl estradiol and progesterone are metabolized through 3A4, so inducers there are going to be a problem. The interaction is the same regardless of the route of delivery: oral, transdermal, vaginal ring, and implant. Now, many of these inducers will only reduce ethinyl estradiol/progesterone by 10%–20%, but the risk is nontrivial and the science inexact, so double protection is a wise strategy here.
TCPR: Inhibitors and inducers are often described as “mild,” “moderate,” or “potent.” What do those terms really mean?
Dr. Sandson: A mild inhibitor will increase blood levels somewhere in the 20%–50% range. Most patients can tolerate that difference, unless they are fragile or elderly. Moderate inhibitors raise levels 60%–100%, and potent inhibitors raise them over 100%, so they more than double it. Tobacco smoking can also be a potent inducer, especially with regard to antipsychotics and some antidepressants, and its effects vary depending on how much one is smoking.
TCPR: What about vaping and e-cigs?
Dr. Sandson: Those are different. It’s actually the polycyclic aromatic hydrocarbons from smoking that induce the enzyme (CYP1A2), and we don’t see that effect with nicotine itself, whether from e-cigs, patch, or gum. We do see some induction with marijuana smoking, which produces the same hydrocarbons. So when patients switch from smoking to e-cigs or quit marijuana, you’d expect to see increased levels of some antipsychotics and antidepressants (Anderson GD et al, Clin Pharmacokinet 2016;55:1353–1358).
Smoking Interactions
TCPR: Do drug interactions start as soon as the medicine is added, or is there a delay?
Dr. Sandson: For inhibitors, the effects typically build up over a few days. Inducers are slower. It takes 2 to 4 weeks for an inducer to rev up and reach full effect, and it’s going to take about that long for reversal of induction. One exception is smoking. It only takes about 3 days for cigarette smoking to become a clinically meaningful inducer, and the reversal of that induction only takes about a week.
TCPR: Going back to inhibitors, are there any that can raise the seizure risk with bupropion?
Dr. Sandson: Possibly. Most antidepressants and antipsychotics can lower the seizure threshold, but bupropion is the biggest offender among the antidepressants available in the US, and the risk goes up with higher levels. Bupropion is metabolized by CYP2B6, and the major inhibitors to worry about adding to it are the “non-pram” SSRIs—that is, all of them except escitalopram and citalopram.
TCPR: What happens when you add those SSRIs to bupropion?
Dr. Sandson: There it gets interesting, because most of bupropion’s norepinephrine reuptake inhibition is accomplished through its metabolite: hydroxybupropion. Drugs that inhibit that conversion, like the non-pram SSRIs, will raise levels of the parent compound and lower levels of the active metabolite. This alters bupropion’s metabolism in a way that effectively creates a new drug I call “Superbutrin.”
TCPR: How does this Superbutrin compare to regular bupropion?
Dr. Sandson: Superbutrin has higher levels of the parent compound, bupropion, and lower levels of the metabolite, hydroxybupropion. In a test tube, the parent compound is twice as potent an inhibitor of norepinephrine reuptake as its metabolite. However, in the body, most of the actual norepinephrine reuptake results from the metabolite because its half-life is much longer than bupropion’s. Everything we know about bupropion—from its seizure risks to its antidepressant effects—flies out the window with Superbutrin. It may be that Superbutrin has a lesser risk of seizures. One study suggests that’s the case, but the data are pretty weak (Silverstone PH, Ann Gen Psych 2008;15(7):19). We really don’t know as much about how this Superbutrin acts.
TCPR: What about drugs that are metabolized through multiple enzymes? Do we have to worry when an inhibitor blocks only one of those enzymes?
Dr. Sandson: Usually not, unless the single pathway that’s inhibited is the major one. That’s true for clozapine, which is predominantly metabolized at CYP1A2, though it has several auxiliary pathways. In general, drugs that are metabolized through only one pathway are more vulnerable to interactions.
TCPR: What are some one-pathway drugs?
Dr. Sandson: We have a few that are pretty exclusive for CYP3A4. Quetiapine (Seroquel), lurasidone (Latuda), and buspirone (Buspar); these are all going to be fairly exclusive at 3A4.
TCPR: Let’s talk about trazodone. It seems like morning sedation is relatively common with that medication.
Dr. Sandson: Yeah, it certainly can be. Trazodone is also a 3A4 substrate, and so giving it with 3A4 inhibitors can heighten and prolong the sedation. But there’s another interaction to worry about with trazodone. One of the metabolites of trazodone, and of nefazodone as well, is something called meta-chlorophenylpiperazine or mCPP, and abrupt increases in mCPP can be both dysphorogenic and anxiogenic, and even hallucinogenic. The body gets rid of mCPP through 2D6, so 2D6 inhibitors can cause it to rise. Potent 2D6 inhibitors include fluoxetine (Prozac), paroxetine (Paxil), and sertraline (Zoloft) at dosages ≥ 150 mg, but many other antidepressants can inhibit it (see table on page 8).
TCPR: Seems like we combine trazodone with those meds a lot. How big a problem is this mCPP?
Dr. Sandson: We don’t see it as often as you’d expect. This interaction tends to happen when trazodone is in the antidepressant dose range, like 300 mg and above. Also, it’s abrupt increases in mCPP that are destabilizing. When chronically administered and slowly titrated, mCPP is actually protective and works as a partial serotonin agonist. So if you start trazodone when a 2D6 inhibitor is already in place, you’ll gradually titrate up the mCPP with the trazodone and there’s no problem. On the other hand, if the patient has been on high-dosage trazodone for a while and a 2D6 inhibitor is suddenly added, then you might expect some adverse psychological effects.
TCPR: What are some of the interactions we should be aware of with the newer antidepressants?
Dr. Sandson: Vortioxetine (Trintellix) is mainly metabolized at CYP2D6, so it’s going to be susceptible to potent inhibitors. For example, bupropion will reliably increase vortioxetine levels about 2-fold. Vilazodone (Viibryd) is a 3A4 substrate, but it’s metabolized by many other enzymes, so it’s actually kind of hard to derange vilazodone levels through inhibition, but 3A4 inducers will drop blood levels. Desvenlafaxine (Pristiq), levomilnacipran (Fetzima), and milnacipran (Savella) are not very susceptible to CYP interactions.
TCPR: You mentioned that drug interactions vary by individual. Can genetic testing clarify that?
Dr. Sandson: Genetic testing tells us about a patient’s enzyme function before any drug interactions. Most people are extensive metabolizers—meaning normal—but at any given enzymatic pathway they can also be “poor,” “intermediate,” or “ultra-rapid.”
TCPR: Do those genetic variations slow or speed up the enzymes like drug interactions do?
Dr. Sandson: The effect is similar, where “poor” would be like having a potent inhibitor on board, “intermediate” a mild-moderate inhibitor, and “ultra-rapid” a potent inducer. But the actual biology is not about fast and slow. An ultra-rapid metabolizer at CYP2D6 doesn’t have faster enzymes, just more of them. Intermediate metabolizers are “slow” because they have fewer enzymes. With poor metabolizers, on the other hand, the issue is one of quality, not quantity: They have a nonfunctional form of the enzyme. A rule of thumb is to cut the dosage in half when dealing with an intermediate metabolizer or a mild-moderate drug interaction, but dosing is difficult and unpredictable with poor or ultra-rapid metabolizers.
TCPR: Another common interaction is lithium and NSAIDs. How strong is that effect?
Dr. Sandson: On average, NSAIDs like ibuprofen and naproxen raise lithium levels 20%–50%, but there’s a wide bell curve here, and it can range from 20% to 200% in practice. Clearly, most cases are going to be in the 20%–50% range. What I do when patients on lithium start an NSAID—other than aspirin or sulindac (Clinoril)—is reduce their lithium level in advance by 30% and recheck a level in a week. Lithium interactions are exceedingly rare with aspirin and sulindac, so I don’t lower the dosage there, but I’ll still check a level after starting them.
TCPR: What’s in the future with drug interactions?
Dr. Sandson: Interactions can happen at many levels: gut absorption, liver metabolism, protein binding, renal excretion, and even the blood-brain barrier. There’s a family of transporters called P-glycoprotein that act like gatekeepers at both the gut lumen and blood-brain barrier, regulating how drugs get into and out of the systemic vasculature and the brain, respectively. When you put it all together it gets rather complicated, and I think what is really needed is better computer software to sort through all this in a meaningful way. We’re not there yet.
TCPR: Thank you for your time, Dr. Sandson.
General Psychiatry Expert Q&ADr. Sandson: The two issues to pay attention to are toxicity and loss of efficacy. Toxicity is the bigger concern, particularly if the drug has a “narrow therapeutic index,” where modest differences in the level can have dangerous effects. Lithium, tricyclics, carbamazepine, and to a lesser extent valproate are some psychiatric examples where high levels can cause problems (see “Toxic Drug Interactions in Psychiatry”). Drug interactions with inhibitors can cause high levels by blocking metabolism by P450 enzymes in the liver. The potency of the inhibitor also matters here. An inhibitor that raises a drug by only 20% is probably not going to cause much concern.
TCPR: What are some potent inhibitors we use?
Dr. Sandson: Fluoxetine (Prozac), fluvoxamine (Luvox), paroxetine (Paxil), and duloxetine (Cymbalta). Fluoxetine and fluvoxamine are potent and hit a broad array of P450 enzymes, while paroxetine’s effects are fairly specific for CYP2D6. Sertraline (Zoloft) is also an inhibitor at CYP2D6, but it doesn’t become potent until you reach the higher dosages, like 150 mg and above.
TCPR: What about inducers?
Dr. Sandson: Inducers do the opposite. They lower drug levels, and potent ones can drop the affected drugs out of the therapeutic range. The inducers often tend to be among the anticonvulsants, and the most potent one we use in psychiatry is carbamazepine (Equetro, Tegretol). Induction is usually not as dangerous as inhibition, but there are exceptions.
TCPR: When is induction a serious problem?
Dr. Sandson: The main examples are oral contraceptives, warfarin, and immunosuppressants (cyclosporins, sirolimus, tacrolimus), all of which are significantly induced by carbamazepine at CYP3A4. Carbamazepine can render birth control ineffective. Lowering immunosuppressants can lead to organ rejection in people who’ve had a transplant, and lowering warfarin can result in ischemic events through coagulation. High levels of warfarin are also dangerous, causing bleeding and hemorrhage, and CYP2C9 inhibitors will raise it.
TCPR: What about other anticonvulsants? Can they induce 3A4 like carbamazepine does?
Dr. Sandson: Other psych meds that induce 3A4 include St. John’s wort, modafinil (Provigil), armodafinil (Nuvigil), topiramate (Topamax), oxcarbazepine (Trileptal), and barbiturates. Several of these are less potent inducers, but you’d still want to avoid them when the risks of induction are serious. The actual interaction may vary by person, as well as by dose. Topiramate and oxcarbazepine’s induction gets more potent as the dosage is increased.
TCPR: Does this 3A4 interaction affect all forms of birth control?
Dr. Sandson: Estrogen and progesterone-based contraceptives will be affected by 3A4 induction, which includes most of them. Ethinyl estradiol and progesterone are metabolized through 3A4, so inducers there are going to be a problem. The interaction is the same regardless of the route of delivery: oral, transdermal, vaginal ring, and implant. Now, many of these inducers will only reduce ethinyl estradiol/progesterone by 10%–20%, but the risk is nontrivial and the science inexact, so double protection is a wise strategy here.
TCPR: Inhibitors and inducers are often described as “mild,” “moderate,” or “potent.” What do those terms really mean?
Dr. Sandson: A mild inhibitor will increase blood levels somewhere in the 20%–50% range. Most patients can tolerate that difference, unless they are fragile or elderly. Moderate inhibitors raise levels 60%–100%, and potent inhibitors raise them over 100%, so they more than double it. Tobacco smoking can also be a potent inducer, especially with regard to antipsychotics and some antidepressants, and its effects vary depending on how much one is smoking.
TCPR: What about vaping and e-cigs?
Dr. Sandson: Those are different. It’s actually the polycyclic aromatic hydrocarbons from smoking that induce the enzyme (CYP1A2), and we don’t see that effect with nicotine itself, whether from e-cigs, patch, or gum. We do see some induction with marijuana smoking, which produces the same hydrocarbons. So when patients switch from smoking to e-cigs or quit marijuana, you’d expect to see increased levels of some antipsychotics and antidepressants (Anderson GD et al, Clin Pharmacokinet 2016;55:1353–1358).
Smoking Interactions
- Antipsychotics
- Olanzapine (Zyprexa), clozapine,
- Haloperidol
- Antidepressants
- Duloxetine (Cymbalta), fluvoxamine (Luvox), mirtazapine (Remeron)
- Other
- Caffeine, propranolol, ramelteon (Rozerem)
Cigarettes and marijuana can lower:
TCPR: Do drug interactions start as soon as the medicine is added, or is there a delay?
Dr. Sandson: For inhibitors, the effects typically build up over a few days. Inducers are slower. It takes 2 to 4 weeks for an inducer to rev up and reach full effect, and it’s going to take about that long for reversal of induction. One exception is smoking. It only takes about 3 days for cigarette smoking to become a clinically meaningful inducer, and the reversal of that induction only takes about a week.
TCPR: Going back to inhibitors, are there any that can raise the seizure risk with bupropion?
Dr. Sandson: Possibly. Most antidepressants and antipsychotics can lower the seizure threshold, but bupropion is the biggest offender among the antidepressants available in the US, and the risk goes up with higher levels. Bupropion is metabolized by CYP2B6, and the major inhibitors to worry about adding to it are the “non-pram” SSRIs—that is, all of them except escitalopram and citalopram.
TCPR: What happens when you add those SSRIs to bupropion?
Dr. Sandson: There it gets interesting, because most of bupropion’s norepinephrine reuptake inhibition is accomplished through its metabolite: hydroxybupropion. Drugs that inhibit that conversion, like the non-pram SSRIs, will raise levels of the parent compound and lower levels of the active metabolite. This alters bupropion’s metabolism in a way that effectively creates a new drug I call “Superbutrin.”
TCPR: How does this Superbutrin compare to regular bupropion?
Dr. Sandson: Superbutrin has higher levels of the parent compound, bupropion, and lower levels of the metabolite, hydroxybupropion. In a test tube, the parent compound is twice as potent an inhibitor of norepinephrine reuptake as its metabolite. However, in the body, most of the actual norepinephrine reuptake results from the metabolite because its half-life is much longer than bupropion’s. Everything we know about bupropion—from its seizure risks to its antidepressant effects—flies out the window with Superbutrin. It may be that Superbutrin has a lesser risk of seizures. One study suggests that’s the case, but the data are pretty weak (Silverstone PH, Ann Gen Psych 2008;15(7):19). We really don’t know as much about how this Superbutrin acts.
TCPR: What about drugs that are metabolized through multiple enzymes? Do we have to worry when an inhibitor blocks only one of those enzymes?
Dr. Sandson: Usually not, unless the single pathway that’s inhibited is the major one. That’s true for clozapine, which is predominantly metabolized at CYP1A2, though it has several auxiliary pathways. In general, drugs that are metabolized through only one pathway are more vulnerable to interactions.
TCPR: What are some one-pathway drugs?
Dr. Sandson: We have a few that are pretty exclusive for CYP3A4. Quetiapine (Seroquel), lurasidone (Latuda), and buspirone (Buspar); these are all going to be fairly exclusive at 3A4.
TCPR: Let’s talk about trazodone. It seems like morning sedation is relatively common with that medication.
Dr. Sandson: Yeah, it certainly can be. Trazodone is also a 3A4 substrate, and so giving it with 3A4 inhibitors can heighten and prolong the sedation. But there’s another interaction to worry about with trazodone. One of the metabolites of trazodone, and of nefazodone as well, is something called meta-chlorophenylpiperazine or mCPP, and abrupt increases in mCPP can be both dysphorogenic and anxiogenic, and even hallucinogenic. The body gets rid of mCPP through 2D6, so 2D6 inhibitors can cause it to rise. Potent 2D6 inhibitors include fluoxetine (Prozac), paroxetine (Paxil), and sertraline (Zoloft) at dosages ≥ 150 mg, but many other antidepressants can inhibit it (see table on page 8).
TCPR: Seems like we combine trazodone with those meds a lot. How big a problem is this mCPP?
Dr. Sandson: We don’t see it as often as you’d expect. This interaction tends to happen when trazodone is in the antidepressant dose range, like 300 mg and above. Also, it’s abrupt increases in mCPP that are destabilizing. When chronically administered and slowly titrated, mCPP is actually protective and works as a partial serotonin agonist. So if you start trazodone when a 2D6 inhibitor is already in place, you’ll gradually titrate up the mCPP with the trazodone and there’s no problem. On the other hand, if the patient has been on high-dosage trazodone for a while and a 2D6 inhibitor is suddenly added, then you might expect some adverse psychological effects.
TCPR: What are some of the interactions we should be aware of with the newer antidepressants?
Dr. Sandson: Vortioxetine (Trintellix) is mainly metabolized at CYP2D6, so it’s going to be susceptible to potent inhibitors. For example, bupropion will reliably increase vortioxetine levels about 2-fold. Vilazodone (Viibryd) is a 3A4 substrate, but it’s metabolized by many other enzymes, so it’s actually kind of hard to derange vilazodone levels through inhibition, but 3A4 inducers will drop blood levels. Desvenlafaxine (Pristiq), levomilnacipran (Fetzima), and milnacipran (Savella) are not very susceptible to CYP interactions.
TCPR: You mentioned that drug interactions vary by individual. Can genetic testing clarify that?
Dr. Sandson: Genetic testing tells us about a patient’s enzyme function before any drug interactions. Most people are extensive metabolizers—meaning normal—but at any given enzymatic pathway they can also be “poor,” “intermediate,” or “ultra-rapid.”
TCPR: Do those genetic variations slow or speed up the enzymes like drug interactions do?
Dr. Sandson: The effect is similar, where “poor” would be like having a potent inhibitor on board, “intermediate” a mild-moderate inhibitor, and “ultra-rapid” a potent inducer. But the actual biology is not about fast and slow. An ultra-rapid metabolizer at CYP2D6 doesn’t have faster enzymes, just more of them. Intermediate metabolizers are “slow” because they have fewer enzymes. With poor metabolizers, on the other hand, the issue is one of quality, not quantity: They have a nonfunctional form of the enzyme. A rule of thumb is to cut the dosage in half when dealing with an intermediate metabolizer or a mild-moderate drug interaction, but dosing is difficult and unpredictable with poor or ultra-rapid metabolizers.
TCPR: Another common interaction is lithium and NSAIDs. How strong is that effect?
Dr. Sandson: On average, NSAIDs like ibuprofen and naproxen raise lithium levels 20%–50%, but there’s a wide bell curve here, and it can range from 20% to 200% in practice. Clearly, most cases are going to be in the 20%–50% range. What I do when patients on lithium start an NSAID—other than aspirin or sulindac (Clinoril)—is reduce their lithium level in advance by 30% and recheck a level in a week. Lithium interactions are exceedingly rare with aspirin and sulindac, so I don’t lower the dosage there, but I’ll still check a level after starting them.
TCPR: What’s in the future with drug interactions?
Dr. Sandson: Interactions can happen at many levels: gut absorption, liver metabolism, protein binding, renal excretion, and even the blood-brain barrier. There’s a family of transporters called P-glycoprotein that act like gatekeepers at both the gut lumen and blood-brain barrier, regulating how drugs get into and out of the systemic vasculature and the brain, respectively. When you put it all together it gets rather complicated, and I think what is really needed is better computer software to sort through all this in a meaningful way. We’re not there yet.
TCPR: Thank you for your time, Dr. Sandson.



Recommended


